Tarea: Cribado Virtual con Smina
Tarea 2: Cribado Virtual con Smina
Objetivo
- Realizar 4 ensayos de cribado virtual (CV) utilizando Smina.
- La librería de moléculas consiste en 50 compuestos del benchmarking set CSAR 2012, el cual incluye compuestos activos (true binders - inhibitors) e inactivos de la proteína CDK2 (experimentalmente probados). Más sobre este challenge CSAR 2012 aquí.
- Se usarán dos conformaciones de la proteína CDK2.
Nota: No descargar los
pdbde las proteínas de estos links. Más abajo se brindan los links de descarga de los archivos a usar.
- Se usarán dos funciones de scoring (a una exhaustividad de 12):
- Cada ensayo de CV combina una conformación de la proteína y una función de scoring. Dándo como resultado 4 ensayos de CV.
- Reportar los resultados y comparar el desempeño del CV en función del scoring utilizado y las conformaciones de la proteína.
Instrucciones
Descarga el archivo de las proteínas y de las moléculas en formato SMILES ⬇️:
- Proteínas :
- Archivo de ligandos (SMILES): cdk2_mols.smi
Descarga los siguientes archivos y guardalos en tu directorio de trabajo ⬇️ (serán necesarios para el análisis final):
Apoyate del siguiente material para realizar los ensayos de CV: Acoplamiento molecular con Smina
Especificaciones de las corridas de docking:
- Usar un valor de exhaustividad de
12. - El sitio activo es el mismo que en la tarea anterior (para ambas conformaciones). Un caja que cubra los residuos 9, 33, 86, 88, 89, 132, 145 y 146 debe ser más que suficiente.
- Usa el parámetro
--scoring vinao--scoring vinardopara definir la función de scoring correspondiente a cada ensayo. Revisa elhelpde smina si tienes dudas (smina --help). - Usa el notebook de jupyter
analisis_resultados_vina.ipynbpara generar las gráficas de resultados de cada ensayo.
- Usar un valor de exhaustividad de
Reporte
En reporte de resultados consiste en un archivo
pdf de cinco diapositivas con lo siguiente:
- Mostrar las 3 gráficas de resultados, debidamente tituladas
y etiquetadas, de cada ensayo de CV en una diapositiva de
ppt:- Gráfico del ranking de moléculas activas vs activas.
- Curva ROC, incluyendo el valor del AUC-ROC.
- Histogramas del scoring de activos vs inactivos.
- En la diapositiva correspondiente, reporta las 3 mejores
moléculas (con menor energía libre de interacción con la proteína) de
cada ensayo de CV.
- Escribe en una tabla el nombre, el valor de energía predicho y si la molécula es realmente activa o no.
En una última diapositiva, responder las siguientes preguntas:
- ¿Consideras que la función de scoring elegida marca alguna diferencia en los resultados del CV? ¿Por qué?
- ¿Consideras que la conformación elegida marca alguna diferencia en los resultados del CV? ¿Por qué?
- ¿Hay alguna diferencia destacable entre los sitios de unión de ambas conformaciones? ¿Cuál?
- Si tuvieras que hacer un cribado virtual para descubrir nuevos inhibidores de la CDK2 a partir de una librería de 1,000,000 de moléculas, ¿Qué conformación y qué función de scoring usarías?
- ¿Qué moléculas activas representan un mayor desafío para el programa de docking? ¿Tendrá algo que ver el tamaño de la molécula?
- En los cuatro ensayos de VS ¿hay moléculas que tiendan a tener siempre los valores más bajos de energía de interacción (scoring)? ¿A qué crees que se deba independientemente de que sean activas o inactivas?
Anota tu nombre en la última diapositiva.
🚨 🚨 🚨 Enviar únicamente el archivo pdf con las cinco diapositivas por mensaje personal a mi cuenta (Joel Ricci) en el Slack del grupo.
📆 Fecha límite de envío es el día sábado de febrero a las 24:00 horas.
Algunos tips:
- 🚨 ¡No te confíes con el tiempo! 🚨
- La duración de cada ensayo de CV puede tomar desde 30 min a 2 horas
⏱ (en promedio 1-1:30 min por molécula) dependiendo de los recursos de
tu equipo y de el número de procesadores que utilices (revisa el
parámetro
cpude smina). - Usa como mínimo dos procesadores (
cpu), si tu equipo cuenta con más, puedes aprovecharlo 💻. - Planifica bien en qué momento realizarás las corridas de docking tomando en cuenta lo anterior 📝.
- La duración de cada ensayo de CV puede tomar desde 30 min a 2 horas
⏱ (en promedio 1-1:30 min por molécula) dependiendo de los recursos de
tu equipo y de el número de procesadores que utilices (revisa el
parámetro
- De preferencia realiza cada ensayo de CV en una carpeta independiente para evitar confundirte y sobreescribir archivos de ensayos previos 😰.
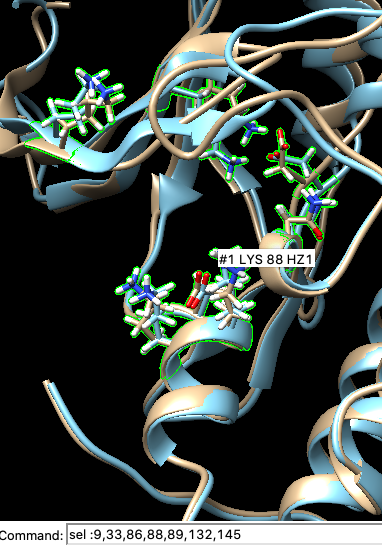
- Recuerda que el tamaño del grid está en Å. Un grid demasiado grande
hará mucho más tardado el análisis. Una caja no mayor a
24x24x24(incluso un poco menos) debería bastar para ambas conformaciones.- En Chimera puedes utilizar el Command Line para
seleccionar los residuos mencionados arriba y usarlos de referencia para
trazar la caja ->
sel :9,33,86,88,89,132,145,146
- En Chimera puedes utilizar el Command Line para
seleccionar los residuos mencionados arriba y usarlos de referencia para
trazar la caja ->
- Basta con hacer la fase de preparación de los
pdbqtuna sola vez. - Si te sirve, anota los comandos y ciclos for que vas utilizando en un archivo de texto (sustituyendo adecuadamente los nombres de los archivos de entrada y salida).
Recursos
- Curso de bash scripting en Datacamp. ¡Tienes acceso a todo el contenido!
- Página de descarga de smina con algunos recursos extra